Стилл реакциясы - Stille reaction
| Стилл реакциясы | |
|---|---|
| Есімімен аталды | Джон Кеннет Стилл |
| Реакция түрі | Ілінісу реакциясы |
| Идентификаторлар | |
| Органикалық химия порталы | стиль-муфта |
| RSC онтологиялық идентификатор | RXNO: 0000035 |
The Стилл реакциясы Бұл химиялық реакция кеңінен қолданылады органикалық синтез. Реакцияға екі органикалық топтың қосылуы жатады, олардың бірі ан түрінде жүзеге асырылады органотинді қосылыс (сонымен бірге органостанналар). Түрлі органикалық электрофилдер басқаларын қамтамасыз етеді байланыстырушы серіктес. Стилл реакциясы - көптеген реакциялардың бірі палладий-катализденген байланыс реакциялары.[1][2][3]
- : Аллил, алкенил, арил, бензил, ацил
- : галогенидтер (Cl, Br, I), псевдогалидтер (OTf, ), OAc
![{ displaystyle { color {Blue} { ce {R ^ {1} -Sn (Alkyl) 3}}} + { color {Red} { ce {R ^ {2} -X}}} { ce {-> [{ color {Green} { ce {Pd ^ {0}}}} { text {(каталитикалық)}}] [{ text {ligand set}}]}} overbrace { { color {Blue} { ce {R ^ {1}}}} ! - ! { color {Red} { ce {R ^ {2}}}}} ^ {coupled product} + { color {Red} { ce {X}}} ! - ! { color {Blue} { ce {Sn (Alkyl) 3}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5baabb66db61c2d31fa2a5ca2b4e8156ee7c4133 "Стилл реакциясының жалпы схемасы")



R1 пробиркилтинге бекітілген топ әдетте sp2- будандастырылған, оның ішінде винил, және арыл топтар.
Бұл органостанндар ауаға да, ылғалға да тұрақты, және осы реактивтердің көпшілігі коммерциялық қол жетімді немесе әдебиет прецедентінен синтезделуі мүмкін. Алайда, бұл қалайы реагенттері өте улы болып келеді. Х әдетте а болып табылады галоид, сияқты Cl, Br, немесе Мен сияқты псевдогалидтер бөлшектер және сульфаттар және фосфаттар пайдалануға болады.[4][5] Бірнеше шолулар жарияланған.[6][2][7][8][9][10][11][12][13][14][15]
Тарих
Бірінші мысал а палладий муфтасы арил галогенидтерінен тұрады органотин реактивтері туралы хабарлады Колин Эборн 1976 ж.[16] Бұл реакциядан диарил өнімі 7% -дан 53% -ға дейін болды. Бұл процесс байланыстыруға дейін кеңейтілді ацилхлоридтер 1977 жылы Тошихико Мигитаның алкил-қалайы реактивтерімен, 53% -дан 87% -ға дейін кетон өнім.[17]

1977 жылы Мигита ілінісу бойынша одан әрі жұмыс жариялады аллил - екеуімен бірге реактивті заттар арыл (C) және ацил (Д.галогенидтер. Аллил топтарының көші-қон қабілеттілігі палладий катализаторы реакцияларды төмен температурада жүргізуге мүмкіндік берді. Арил галогенидтерінің шығымы 4% -дан 100% -ке дейін, ал ацил галогенидтер үшін 27% -дан 86% -ға дейін болды.[18][19] Мигита мен Косугидің алғашқы үлестерін көрсете отырып, Стилл реакциясы кейде деп аталады Мигита - Косуги - Стилл байланысы.

Стилл кейіннен алкил қалайы реагенттерінің көптеген арил және ацил галогенидтерімен көптеген реакциялар жағдайында (76% -99%) жұмсақ реакция жағдайында байланысқандығы туралы хабарлады.[18][20] Стилл 1980 жылдары осы кең және жұмсақ процесті қолдана отырып, көптеген кетондарды синтездеу бойынша жұмысын жалғастырды және осы трансформация механизмін түсіндірді.[21][22]

1980 жылдардың ортасына қарай, реакцияның субстрат аясын зерттеуді жалғастыра отырып, қалайы қатысатын реакциялар тақырыбында 65-тен астам мақалалар жарық көрді. Осы саладағы алғашқы зерттеулер алкил топтарының түйісуіне бағытталса, болашақ жұмыстардың көп бөлігі синтетикалық жағынан пайдалы байланыстыруды қамтыды винил, алкенил, арил және аллил органостанналар галогенидтерге дейін. Осы органотин реактивінің ауаға тұрақтылығы және олардың синтезделуінің жеңілдігі арқасында Стилл реакциясы органикалық синтезде кең таралды.[8]
Механизм
Стилл реакциясының механизмі жан-жақты зерттелген.[11][23] The каталитикалық цикл қамтиды тотықтырғыш қосу а галоид немесе псевдогалид (2) а палладий катализаторы (1), трансметалдау туралы 3 бірге органотин реактиві (4), және редуктивті жою туралы 5 біріктірілген өнімді беру үшін (7) және қалпына келтірілген палладий катализаторы (1).[24]

Алайда, Stille байланысының егжей-тегжейлі механизмі өте күрделі және көптеген реакция жолдары арқылы жүруі мүмкін. Басқалар сияқты палладий-катализденген байланыс реакциялары, белсенді палладий катализаторы 14 электронды Pd (0) кешені болып саналады, оны әр түрлі жолмен жасауға болады. 18 немесе 16 электронды Pd (0) Pd (PPh) көзін пайдалану3)4, Pd (дба)2 өтуі мүмкін лиганд белсенді түрлерді қалыптастыру үшін диссоциация. Екіншіден, фосфиндер лигандсыз палладийге қосуға болады (0). Соңында, суреттегідей, төмендету Pd (II) көзінен (8) (Pd (OAc)2, PdCl2(MeCN)2, PdCl2(PPh3)2, BnPdCl (PPh3)2және т.б.) фосфинді лигандтармен немесе органотинді реактивтермен қосылады [6]
Тотықтырғыш қоспа
14 электронды Pd (0) кешеніне тотықтырғыш қосу ұсынылады. Бұл процесс 16 электронды Pd (II) түрін береді. Анионды деген болжам жасалды лигандтар, сияқты OAc, [Pd (OAc) (PR) түзілуімен осы қадамды жылдамдатыңыз3)n]−, палладий түрлерін көбірек нуклеофилді етеді.[11][25]Кейбір жағдайларда, әсіресе sp3- будандастырылған органогалид қолданылады, an SN2 типті механизм басымдыққа ұмтылады, дегенмен бұл әдебиетте жиі кездеседі.[11][25]Алайда, қалыпты қалыптасқанына қарамастан cisа. аралық келісілген тотығу қоспасы, бұл өнім жылдам тепе-теңдік онымен транс-исомер.[26][27]

Трансметалдау
The трансметалдау туралы транс аралық тотықтырғыш қосу қадам субстраттар мен жағдайларға байланысты әр түрлі механизмдер арқылы жүреді деп саналады. Stille муфтасына арналған трансметаляцияның ең көп таралған түрі ан ассоциативті механизм. Бұл жол білдіреді органостаннан, әдетте а қалайы аллил, алкенил немесе арил тобымен байланысқан атом мүмкін үйлестіру палладийге осы қос байланыстың бірі арқылы. Бұл өткінші бес валент шығарады, 18 электронды түрлер, содан кейін а түзу үшін лиганд отрядынан өтуі мүмкін шаршы жазықтық қайтадан күрделі. Палладийге R арқылы үйлестірілген органостананға қарамастан2 топ, R2 ресми түрде палладий (R2-Sn байланысы үзілуі керек), ал X тобы трансметаляцияны аяқтай отырып, қалайымен бірге кетуі керек. Бұл екі механизм арқылы жүреді деп есептеледі.[28]
Біріншіден, қашан органостаннан бастапқыда транс металлдар кешеніне қосылады, Х тобы мүмкін үйлестіру дейін қалайы, палладийден басқа, циклды шығарады өтпелі мемлекет. Осы қосылыстың бұзылуы R жоғалтуға әкеледі3Sn-X және үш валентті палладий R-мен бірге1 және Р.2 а cis қарым-қатынас. Басқа жиі кездесетін механизмге органостаннанның бастапқы қосылысы жатады транс жоғарыда көрсетілгендей палладий кешені; дегенмен, бұл жағдайда Х тобы қалайыға координат жасамайды, саңылау шығарады өтпелі мемлекет. Кейін α-көміртегі Палладийге қалайының шабуылына қатысты, қалайы кешені оң зарядпен кетеді. Төмендегі схемада қалайыға үйлестіретін қос байланыс R болатынын ескеріңіз2, сондықтан кез келген алкенил, аллил, немесе арыл топ. Сонымен қатар, X тобы механизм кез келген уақытта диссоциацияланып, Sn-мен байланысуы мүмкін+ соңында кешенді. Тығыздықтың функционалдық теориясы есептеулер, егер 2 ашық механизм басым болады деп болжайды лигандтар палладиймен байланысқан күйінде қалады және X тобы жапырақтары, ал циклдік механизм ықтималдығы лиганд диссоциацияланғанға дейін трансметалдау. Демек, полярлы еріткіштердегі ұсақ бөлшектер сияқты жақсы кету топтары біріншісіне, ал әзірге жағымды фосфинді ірі лигандтар соңғысына артықшылық береді.[28]

Үшін аз таралған жол трансметалдау арқылы диссоциативті немесе еріткіштің көмегімен механизм. Мұнда тетравалентті палладий түрлерінен лиганд диссоциацияланады, ал координациялық еріткіш палладийге қосыла алады. Қашан еріткіш ажыратады, 14 электронды үш валентті аралықты құру үшін органостаннан қосуға болады палладий, жоғарыдағыдай ашық немесе циклдік типтегі процестен өту.[28]
Редуктивті жою сатысы
R үшін1-Р2 дейін редуктивті жою, бұл топтар өзара орналасуы керек cis үйлестіру алаңдары. Кез келген транс-деміргіштер сондықтан изомерленуі керек cis аралық немесе муфта көңілсіз болады. Редуктивті жоюдың түрлі механизмдері бар және олар әдетте келісілген болып саналады.[11][29][30]
Біріншіден, 16 электронды төрт валентті аралық трансметалдау қадам а-дан көмекші емес редуктивті жоюға ұшырауы мүмкін шаршы жазықтық күрделі. Бұл реакция екі сатыда жүреді: біріншіден, редуктивті элиминация R-мен жаңадан пайда болған сигма байланысының координациясымен жалғасады1 және Р.2 байланыстырылған өнімді беретін соңғы диссоциациямен металға.[11][29][30]

Алдыңғы процесс кейде баяу жүреді және 14-электронды алу үшін лигандтың диссоциациялануымен едәуір жылдамдауы мүмкін. T пішінді аралық. Содан кейін бұл аралық Y-тәрізді қоспа түзе алады, ол тезірек редуктивті элиминациядан өтуі мүмкін.[11][29][30]

Ақырында, қосымша лиганд палладиймен байланысып, 18 электронды тригональды бипирамидалық құрылым түзе алады, R1 және Р.2 экваторлық позицияларда бір-біріне cis. Бұл аралықтың геометриясы оны жоғарыдағы Y-тәрізді етіп жасайды.[11][29][30]

Болуы ірі лигандтар сонымен қатар жою жылдамдығын арттыра алады. Сияқты лигандар фофиндер үлкенмен шағу бұрыштары себеп стерикалық репульсия L мен R аралығында1 және Р.2, нәтижесінде L мен R топтары арасындағы бұрыш өседі және R арасындағы бұрыш пайда болады1 және Р.2 сондықтан тезірек мүмкіндік беретін төмендеу редуктивті жою.[11][24]

Кинетика
Организаторлардың жылдамдығы трансметалатты палладий катализаторларымен төменде көрсетілген. Sp2-қаңылтырға байланған гибридтенген көміртегі топтары ең көп қолданылатын байланыстырушы серіктес болып табылады және sp3- будандастырылған көміртектер қатал жағдайларды қажет етеді және терминал алкиндер C-H байланысы арқылы қосылуы мүмкін Соногашираның реакциясы.

Органикалық қалайы қосылысы ретінде әдетте триметилстаннил немесе трибутилстаннил қосылысы қолданылады. Триметилстаннил қосылыстары трибутилстаннил қосылыстарымен салыстырғанда жоғары реактивтілік көрсетеді және әлдеқайда қарапайым 1H-NMR спектрлері, біріншісінің уыттылығы әлдеқайда көп.[31]
Қандай лигандтардың реакциясы жоғары өнімділікпен және айналым жылдамдығымен жақсы жүретіндігін оңтайландыру қиынға соғады. Себебі тотықтырғыш қосу электрондарға бай металды қажет етеді, демек, лигандтарды электронды түрде береді. Алайда, электрондардың жетіспейтін металы үшін қолайлы трансметалдау және редуктивті жою қадамдар, мұнда электронды алып тастайтын лигандтар жақсы. Сондықтан оңтайлы лиганд жиынтығы жеке субстраттар мен қолданылатын жағдайларға байланысты. Бұл жылдамдықты анықтайтын қадамды, сондай-ақ механизмді өзгерте алады трансметалдау қадам.[32]
Әдетте, фосфиндер сияқты аралық донорлық лигандтар қолданылады. Қарқынды жоғарылатуды орташа электронға бай лигандтар, мысалы, три-2-фурилфосфин немесе трифениларсенин қолданған кезде байқауға болады. Сол сияқты донорлар саны жоғары лигандалар байланысу реакцияларын бәсеңдетуі немесе тежеуі мүмкін.[32][33]
Бұл бақылаулар, әдетте, Стилл реакциясы үшін жылдамдықты анықтайтын қадам болып табылады трансметалдау.[33]
Қоспалар
Стилл реакциясының ең көп таралған қоспасы болып табылады стехиометриялық немесе ко-каталитикалық мыс (I), нақты мыс йодиді жақсарта алады ставкалар > 10-ға дейін3 бүктеу Бұл полярлық деп тұжырымдалған еріткіштер мыс трансметалатты бірге органостаннан. Нәтижесінде органокупрат реактив палладий катализаторымен трансметалдауы мүмкін. Сонымен қатар, эфирлік еріткіштерде мыс а-ны кетіруді жеңілдетуі мүмкін фосфинді лиганд, Pd орталығын қосу.[9][34][35][36][37]
Литий хлориді X тобы палладийден бөлінетін (мысалы, ашық механизм) жағдайында жылдамдықтың жылдамдатқышы екені анықталды. The хлорид ион катализаторды неғұрлым белсенді ететін палладийдегі X тобын ығыстырады деп саналады трансметалдау немесе жылдамдығын арттыру үшін Pd (0) қосымшасына үйлестіру арқылы тотықтырғыш қосу. Сондай-ақ, LiCl тұзы жақсартуды жақсартады полярлық бұл әдетте анионды жеңілдететін еріткіш лиганд (–Cl, –Br, –OTf кету. Бұл қоспа еріткіштің қажет кезінде қажет THF қолданылады; дегенмен, неғұрлым полярлы еріткішті қолдану NMP, осы тұз қоспасына деген қажеттілікті алмастыра алады. Алайда, муфтаның трансметалдау қадамы циклдік механизм арқылы жүрсе, литий хлоридін қосу жылдамдықты төмендетуі мүмкін. Циклдік механизмдегідей, бейтарап лиганд, мысалы, фосфин, аниондық Х тобының орнына диссоциациялануы керек.[10][38]
Соңында фтор иондары, сияқты фторлы цезий, сонымен қатар әсер етеді каталитикалық цикл. Біріншіден, фтор реакцияларының жылдамдығын арттыра алады органотрифлаттар, мүмкін сол сияқты әсер етуі мүмкін литий хлориді. Сонымен қатар, фтор иондары әрекет ете алады қоқыс жинаушылар үшін қалайы қосалқы өнімдер арқылы жоюды жеңілдетеді сүзу.[36]
Бәсекелес жанама реакциялар
Стилл реакциясымен байланысты ең көп таралған жанама реактивтілік - бұл станан реактивтерін R түзу үшін біртектес біріктіру.2-Р2 күңгірт. Екі мүмкін механизм арқылы жүреді деп саналады. Біріншіден, -ның екі эквивалентінің реакциясы органостаннан Pd (II) прекатализаторынан кейін біртектес өнім шығады редуктивті жою. Екіншіден, Pd (0) катализаторы a өтуі мүмкін радикалды процесс димерді беру үшін. Қолданылатын органостаннан реагенті әдеттегідей қаңылтырда төрт валентті, әдетте сп-тен тұрады2- ауыстырылатын топ және үш «ауыстырылмайтын» алкил топтар. Жоғарыда көрсетілгендей, алкил топтары палладий катализаторына қоныс аударғанда ең баяу жүреді.[10]

Сонымен қатар, 50 ° C-тан төмен температурада, арыл екеуінде де топтар палладий және а үйлестірілген фосфин айырбастауға болады. Әдетте анықталмағанымен, олар көптеген жағдайларда ықтимал кішігірім өнім бола алады.[10]
Сонымен, сирек кездесетін және экзотикалық жанама реакция ретінде белгілі киноны ауыстыру. Мұнда, бастапқыдан кейін тотықтырғыш қосу туралы арил галогенид, бұл Pd-Ar түрі винил қаңылтыр қос байланысын ендіре алады. Кейін β-гидридті жою, қоныс аудару және протестаннеляция синтезделуі мүмкін, 1,2-бөлінген олефин.[10]

Көптеген басқа жанама реакциялар болуы мүмкін және оларға жатады E / Z изомеризациясы, алкенилстаннан қолданған кезде проблема туындауы мүмкін. Бұл трансформацияның механизмі қазіргі кезде белгісіз. Қалыпты, органостанналар үшін өте тұрақты гидролиз, бірақ өте электрондарға бай арил станналары қолданылған кезде, бұл айтарлықтай реакцияға айналуы мүмкін.[10]
Қолдану аясы
Электрофил
Винил галогенидтері Стилл реакциясының байланыстырушы серіктестері болып табылады және осы типтегі реакциялар көптеген кездеседі табиғи өнім жалпы синтездер. Әдетте винил иодидтер мен бромидтер қолданылады. Винилхлоридтер реактивті емес тотықтырғыш қосу Pd (0) дейін. Йодидтер әдетте артықшылықты: олар әдетте жылдамырақ және жұмсақ жағдайда реакция жасайды бромидтер. Бұл айырмашылық төменде селективті түрде көрсетілген муфта винил бромидінің қатысуымен винил иодиді.[10]
Әдетте стереохимия туралы алкен реакцияның қатал жағдайларын қоспағанда, бүкіл реакция кезінде сақталады. Әр түрлі алкендер қолданылуы мүмкін, оларға α- және β-гало-α, β қанықпаған жатады. кетондар, күрделі эфирлер, және сульфоксидтер (әдетте, мыс (I) қоспасы қажет) және басқалары (төмендегі мысалды қараңыз).[39] Кейде винилді трифлеттер де қолданылады. Кейбір реакцияларға қосуды қажет етеді LiCl және басқалары баяулады, бұл екі механикалық жол бар дегенді білдіреді.[10]

Жалпыға ортақ тағы бір класс электрофилдер арыл және гетероциклді галогенидтер. Винилді субстраттарға келетін болсақ, бромидтер мен йодидтер көп шығындарға қарамастан жиі кездеседі. Электронды донорлық алмастырғыштармен алмастырылған сақиналарды қоса, көптеген арил топтарын таңдауға болады, биарил сақиналар және т.б. Галогенмен алмастырылған гетероциклдар байланыстырушы серіктестер ретінде де қолданылды, оның ішінде пиридиндер, фурандар, тиофендер, тиазолалар, indoles, имидазолдар, пуриндер, урацил, цитозиндер, пиримидиндер, және тағы басқалары (Гетероциклдер кестесін төменде қараңыз; галогендерді әрқайсысында әртүрлі күйде ауыстыруға болады).[10]

Төменде күрделілікті арттыру үшін Stille муфтасын қолданудың мысалы келтірілген гетероциклдар туралы нуклеозидтер, сияқты пуриндер.[40]

Арыл бөлшектер және сульфаттар органостаннан реактивтерінің алуан түрлілігі. Трифлеттер Стиль реакциясындағы бромидтермен салыстырмалы түрде әрекет етеді.[10]
Ацилхлоридтер байланыстырушы серіктестер ретінде де қолданылады және оларды алу үшін органостаннанның, алкил-қалайы реагенттерінің үлкен спектрімен қолдануға болады кетондар (төмендегі мысалды қараңыз).[41] Алайда кейде ацилхлоридті енгізу қиынға соғады функционалдық топтар сезімтал функционалды топтары бар үлкен молекулаларға. Бұл үдеріске жасалған альтернатива - стиль-карбонилитті тоғысу реакциясы карбонил арқылы топ көміртегі тотығын енгізу.[10]

Аллил, бензилді, және пропаргилик галогенидтерді қосуға болады. Әдетте аллилогенді галогенидтер employed арқылы жүреді3 органостаннанмен α немесе γ күйінде қосылуға мүмкіндік беретін өтпелі күй, ең аз алмастырылған көміртекте пайда болады (төмендегі мысалды қараңыз).[42] Алкенил эпоксидтері (іргелес) эпоксидтер және алкендер ) сондай-ақ through арқылы осы жұптасудан өтуі мүмкін3 өтпелі мемлекет сияқты, эпоксидті ан алкоголь. Аллилді және бензилді ацетаттар әдетте қолданылады, пропаргиликалық ацетаттар органостанандармен реактивті емес.[10]

Станнейн
Органостаннан реактивтері жалпы болып табылады. Бірнеше коммерциялық қол жетімді.[43] Станнан реактивтерін а реакциясы арқылы синтездеуге болады Григнард немесе органолитий реактиві пробиркилтин хлоридтерімен. Мысалға, винилтрибутилтин бромды винилмагнийдің трибутилтин хлоридімен әрекеттесуі арқылы дайындалады.[44] Гидростанилдану туралы алкиндер немесе алкендер көптеген туындыларды ұсынады. Органотин реактивтері ауа мен ылғалға төзімді. Кейбір реакциялар суда да жүруі мүмкін.[45] Оларды тазартуға болады хроматография. Олар көптеген функционалды топтарға төзімді. Кейбір органотинді қосылыстар өте көп улы, әсіресе триметилстаннил туындылары.[10]
Винилстаннан немесе алкенилстаннан реактивтерін қолдану кең таралған.[10] Шектеулерге қатысты өте үлкен көлемді станан реактивтері де, олардың орнына алмастырғыштар да бар α-көміртегі баяу әрекет етеді немесе оңтайландыруды талап етеді. Мысалы, төмендегі жағдайда, α-алмастырылған винилстаннан тек терминалды йодидпен реакцияға түседі стерикалық кедергі.[46]
Арыстанстан реактивтері де кең таралған және екеуі де электронды донорлық және электронды алу топтар трансметалдау жылдамдығын іс жүзінде жоғарылатады. Бұл тағы екі механизмді білдіреді трансметалдау орын алуы мүмкін. Бұл реагенттердің жалғыз шектеуі - орто-позициядағы орынбасарлар, өйткені метил топтары реакция жылдамдығын төмендетуі мүмкін. Түрлі гетероциклдар (Электрофил бөлімін қараңыз) байланыстырушы серіктес ретінде де қолданыла алады (мысалға тиазол төменде қоңырау).[10][47]


Станилдердің ішіндегі ең реактивті алькинилстаннаналар Стилл муфталарында да қолданылған. Олар әдетте қажет емес, өйткені терминал алкиндері палладий катализаторларына өздерінің C-H байланысы арқылы тікелей қосыла алады Соногашира байланысы. Аллилстаннаналар жұмыс істеді, бірақ аллилогеноидтар сияқты бақылау қиынға соғады. региоселективтілік α және γ қосу үшін. Дистаннан және ацил станнан реактивтері Стилл муфталарында да қолданылған.[10]
Қолданбалар
Стилл реакциясы әртүрлі полимерлердің синтезінде қолданылған.[48][49][50] Алайда, Стилл реакциясының ең кең қолданылуы - бұл қолдану органикалық синтездер, және, атап айтқанда, синтезінде табиғи өнімдер.
Табиғи өнімнің жалпы синтезі
Овермендікі 19-қадам энантиоселективті жалпы синтез квадригеминнің құрамында қос стиль бар айқас метатеза реакция.[6][51] Күрделі органостаннан екі арил-йодидтер тобына қосылады. Екі еселенгеннен кейін Гек циклизация, өнімге қол жеткізіледі.

Панектің 32 қадамы энантиоселективті жалпы синтез туралы ансамицин антибиотик (+) - микотриенол Still типіндегі макроциклдің соңғы сатыдағы тандемін қолданады. Мұнда органостанның алкенге шабуылдаған екі трибутил қалайы тобы бар. Бұл органостаннан сызықтық бастапқы материалдың екі ұшын макроциклге «жабыстырады», процесте жетіспейтін екі метилен қондырғысын қосады. Хош иісті өзегін тотықтырғаннан кейін қышқыл аммоний нитраты (БОЛАДЫ) және қорғаныстан шығару бірге фторлы қышқыл табиғи өнімді 3 қадамнан 54% өнім береді.[6][52]

Стивен Мартин және әріптестерінің 21 сатылы энтамиэлективті манцаминді антитуморлық алкалоидты ирцинал А-тің жалпы синтезі бір кастрюльді Стиль / Дильс-Алдер реакциясын қолданады. Алкин тобы бромды винилге қосылады, содан кейін ан орнында Дильс-Алдер циклдік шығарылым қосылған алкен мен алкеннің арасында пирролидин сақина.[6][53]

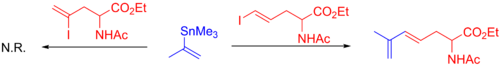
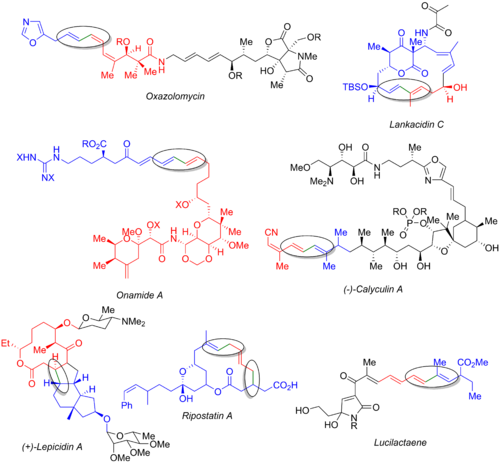
Стилл реакциясын көптеген басқа синтездер, соның ішінде оксазоломициннің реакциясын пайдаланады,[54] ланкацидин С,[55] онамид А,[56] кальцулин А,[57] лепицидин А,[58] рипостатин А,[59] және лукилактаен.[6][60] Төмендегі сурет финалды көрсетеді табиғи өнім, органогалид (көк), органостаннан (қызыл) және түзіліп жатқан байланыс (жасыл және шеңбер). Осы мысалдардан Стилл реакциясын синтездің бастапқы кезеңінде де (оксазоломицин және кальцулин А), конвергентті жолдың соңында (онамид А, ланкацидин С, рипостатин А), немесе орта (лепицидин А және люцилактаен). Рипостатин А синтезінде екі қатар жүретін Стилл муфталары, содан кейін а сақинаны жабатын метатеза. Люцилактаеннің синтезінде орташа суббірлік бар, оның бір жағында бор, екінші жағында станнан бар, бұл кейіннен Сузукидің ілінісуімен Стилл реакциясын жүргізуге мүмкіндік береді.
Вариациялар
Әр түрлі органикалық еріткіштерде реакцияны жүргізуден басқа, суда еріткіште Стилл муфталарының кең спектрін алуға мүмкіндік беретін жағдайлар жасалған.[14]
Cu (I) тұздарының қатысуымен, палладий-көміртегі тиімді катализатор ретінде көрсетілген.[61][62]
Саласында жасыл химия сияқты Стилл реакциясы қанттың аз балқитын және жоғары полярлы қоспасында жүретіні туралы хабарланған маннит, а мочевина сияқты диметилмочевина және тұз аммоний хлориді[63].[64] Катализатор жүйесі трис (дибензилиденацетон) дипалладий (0) бірге трифениларсин:

Стиль - карбонилитті айқаспалы муфталар
Стиль муфтасының жалпы өзгерісі - а карбонил R арасындағы топ1 және Р.2, қалыптастырудың тиімді әдісі ретінде қызмет етеді кетондар. Бұл процесс Мичита мен. Алғашқы барлау жұмыстарына өте ұқсас Стилл (Тарихты қараңыз) органостаннан байланыстыру ацилхлоридтер. Алайда, бұл бөліктер әрдайым қол жетімді бола бермейді және оларды қалыптастыру, әсіресе сезімталдардың қатысуымен болуы мүмкін функционалдық топтар. Сонымен қатар, олардың жоғары реактивтілігін бақылау қиынға соғуы мүмкін. Стилл-карбонилді кросс муфтасы атмосфераны қоспағанда, Stille муфтасымен бірдей шарттарды қолданады. көміртегі тотығы (CO) пайдаланылуда. CO палладий катализаторымен үйлестіре алады (9) алғашқы тотықтырғыш қосудан кейін, содан кейін СО енгізу Pd-R ішіне1 облигация (10) нәтижесінде пайда болады редуктивті жою кетонға (12). The трансметалдау қадам әдетте болып табылады ставканы анықтайтын қадам.[6]

Ларри Овермен және әріптестер өздерінің 20 сатысында Stille-карбонилиттік кросс муфтасын пайдаланады энантиоселективті жалпы синтез туралы стрихнин. Қосылған карбонил кейіннен а арқылы терминалды алкенге айналады Виттиг реакциясы негізгі азот пен пентациклді ядро аза- арқылы түзілуіне мүмкіндік береді.Қиындық -Маннич реакциясы.[6][65]
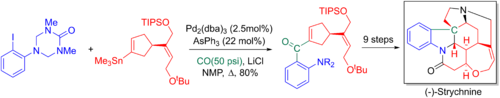
Джорджио Ортар және басқалар. синтездеу үшін Stille-карбонилиттік кросс муфтасын қалай қолдануға болатындығын зерттеді бензофенон фосфор. Олар 4-бензоил-L-фенилаланинге енгізілген пептидтер және әртүрлі пептидті-ақуызды өзара әрекеттесуді зерттеу үшін олардың фотоафинділік белгілері үшін қолданылады.[6][66]

Луи Хегедустың 16 қадамы рацемиялық жалпы синтез Джатрафонның 11 мүшені қалыптастыру үшін соңғы сатысы ретінде стилл-карбонилиттік кросс муфтасы болды макроцикл. Хайлда орнына винил трифлеті байланыстырушы серіктес ретінде қолданылады.[6][67]

Стилл мен Келлидің байланысы
Жарияланған басылымды қолдану арқылы Eaborn 1976 жылы арилгалидтерден және дистаннаннан арилстаннан түзетін Т.Росс Келли бұл процесті молекулалық арилгалидтердің қосылуы. Бұл тандемдік стенниляция / арил галогенді байланысы әртүрлі дигидрофенантрендердің синтезі үшін қолданылған. Құрылған ішкі сақиналардың көпшілігі 5 немесе 6 мүшелермен шектеледі, дегенмен макроциклизацияның кейбір жағдайлары туралы хабарланған. Кәдімгі Стилл муфтасынан айырмашылығы, хлор галоген ретінде жұмыс істемейді, мүмкін оның реактивтілігі төмен болғандықтан галоген реттілік (оның байланыс ұзындығы неғұрлым қысқа және байланыстың күшті диссоциациялану энергиясы арқылы өтуді қиындатады тотықтырғыш қосу ). Төмендегі схеманың ортасынан бастап және сағат тілімен жүру арқылы палладий катализаторы (1) тотықтырғыш қосады ең реактивті C-X байланысына дейін (13) қалыптастыру 14, ілесуші трансметалдау дистаннанмен (15) өнім беру 16 және редуктивті жою арилстаннан алу үшін (18). Қалпына келтірілген палладий катализаторы (1) мүмкін тотықтырғыш қосыңыз екінші C-X байланысына 18 қалыптастыру 19, содан кейін молекулалық трансметалдау өнім беру 20, ілесуші редуктивті жою біріктірілген өнімді беру үшін (22).[6]

Дже Джек Ли және басқалар. Стиль-Келли муфтасын әр түрлі бензолардың синтезінде қолданды [4,5] фуропиридиндердің сақина жүйелерін. Олар а-ны қамтитын үш сатылы процесті қолданады Бухвальд-Хартвиг, басқа палладий-катализденген байланыс реакциясы, содан кейін молекулааралық Стилл-Келли ілінісі. Арил-йодидті байланыс болатынын ескеріңіз тотықтырғыш қосыңыз дейін палладий арил-бромды байланыстың екеуіне қарағанда жылдамырақ.[6][68]
![Бензоның синтезі [4,5] фуропиридиндер](http://upload.wikimedia.org/wikipedia/commons/thumb/2/25/Benzofuropyridines.png/500px-Benzofuropyridines.png)
Сондай-ақ қараңыз
- Органотинді химия
- Органостаннан қоспа
- Палладий-катализденген байланыс реакциялары
- Сузуки реакциясы
- Негиши муфтасы
- Гек реакциясы
- Хияма ілінісі
Әдебиеттер тізімі
- ^ Хартвиг, Дж. Ф. Органотрансформациялық металдар химиясы, байланыстырудан катализге дейін; Университеттің ғылыми кітаптары: Нью-Йорк, 2010. ISBN 189138953X
- ^ а б Стилл, Дж. К. Angew. Хим. Int. Ред. Энгл. 1986, 25, 508–524. (Шолу )
- ^ Фарина, V .; Кришнамурти, V .; Скотт, В. Дж. Org. Реакция. 1998, 50, 1–652. (Шолу )
- ^ Скотт, В.Дж .; Қытырлақ, Г.Т .; Стилл, Дж. К. Органикалық синтез, Coll. Том. 8, б. 97 (1993); Том. 68, б. 116 (1990). (Мақала )
- ^ Стилл, Дж. К .; Эчаваррен, А.М .; Уильямс, Р.М .; Гендрикс, Дж. А. Органикалық синтез, Coll. Том. 9, б.553 (1998); Том. 71, с.97 (1993). (Мақала )
- ^ а б c г. e f ж сағ мен j к л Курти, Л .; Чако, Б. Органикалық синтездегі реакциялардың стратегиялық қолданылуы; Elsevier: Берлингтон, 2005 ж.
- ^ Митчелл, Т. J. Organomet. Хим., 1986, 304, 1-16.
- ^ а б Митчелл, Т. Синтез, 1992, 803-815. (дои:10.1055 / с-1992-26230 )
- ^ а б Фарина, В. Таза Appl. Хим., 1996, 68, 73–78. (дои:10.1351 / pac199668010073 ).
- ^ а б c г. e f ж сағ мен j к л м n o б Фарина, V .; Кришнамурти, V .; Скотт, В. Дж. Стилл реакциясы; Вили: Онлайн, 2004. (дои:10.1002 / 0471264180.немесе050.01 ).
- ^ а б c г. e f ж сағ мен Эспинет, П .; Эчаваррен, А.М. Angew. Хим. Int. Ред., 2004, 43, 4704–4734.(дои:10.1002 / anie.200300638 )
- ^ Паттенден, Г .; Синклер, Д. Дж. Дж. Органомет. Хим., 2002, 653, 261-268.
- ^ Косуги М .; Фугами, К. J. Organomet. Хим., 2002, 19, 10-16.
- ^ а б Пьер Дженет, Дж .; Савиньяк, М. J. Organomet. Хим., 1999, 576, 305-317.
- ^ Кордова, С .; Бартоломе, С .; Мартинес-Илардуя, Дж.М ..; Эспинет, П. ACS Catal., 2015, 5, 3040–3053.(дои:10.1021 / acscatal.5b00448 ).
- ^ Азариан, Д .; Дуа, С.С .; Эборн, С .; Уолтон, Д. J. Organomet. Хим., 1976, 117, C55-C57. (дои:10.1016 / S0022-328X (00) 91902-8 )
- ^ Косуги М .; Шимизу, Ю .; Мигита, Т. Хим. Летт., 1977, 6, 1423-1424. (дои:10.1246 / cl.1977.1423 )
- ^ а б Косуги М .; Сасазава, К .; Шикизу, Ю .; Мигита, Т. Хим. Летт., 1977, 6, 301-302. (дои:10.1246 / cl.1977.301 )
- ^ Косуги М .; Шимизу, Ю .; Мигита, Т. J. Organomet. Хим., 1977, 129, C36-C38. (дои:10.1016 / S0022-328X (00) 92505-1 )
- ^ Милштейн, Д .; Стилл, Дж. К. Дж. Хим. Soc., 1978, 100, 3636-3638. (дои:10.1021 / ja00479a077 )
- ^ Милштейн, Д .; Стилл, Дж. К. Дж. Хим. Soc., 1979, 101, 4992-4998. (дои:10.1021 / ja00511a032 )
- ^ Милштейн, Д .; Стилл, Дж. К. Дж. Орг. Хим., 1979, 44, 1613-1618. (дои:10.1021 / jo01324a006 )
- ^ Касадо, А.Л .; Эспинет, П .; Галлего, А.М. Дж. Ам, Хим. Soc., 2000, 122, 11771-11782. (дои:10.1021 / ja001511o )
- ^ а б Crabtree, R. H. Өтпелі металдардың металлорганикалық химиясы, 5-ші басылым; Вили: Нью-Йорк, 2009 ж.
- ^ а б Перес-Темпрано, М. Х .; Галлего, А.М .; Касарес, Дж. А .; Эспинет, П. Органометалл, 2011, 30, 611-617. (дои:10.1021 / om100978w ).
- ^ Миннити, Д. Инорг. Хим, 1994, 33, 2631-2634.(дои:10.1021 / ic00090a025 ).
- ^ Касадо, А.Л .; Эспинет, П. Органометалл, 1998, 17, 954-959. (дои:10.1021 / om9709502 ).
- ^ а б c Гарсия-Мельчор, М .; Брага, A. A. C .; Лледос, А .; Уякью, Г .; Масерас, Ф. Acc. Хим. Res., 2013, 46, 2626-2634. (дои:10.1021 / ar400080r )
- ^ а б c г. Джилли, А .; Стилл, Дж. К. Дж. Хим. Soc., 1980, 102, 4933-4941. (дои:10.1021 / ja00535a018 ).
- ^ а б c г. Браун, Дж. М .; Кули, Н. Хим. Аян, 1988, 88, 1031-1046. (дои:10.1021 / cr00089a003 ).
- ^ Маккиллоп, А .; Абель, Е. В .; Stone, F. G. A .; Уилкинсон, Г. Органометалл химиясы II, Elsevier Scientific: Оксфорд, 1995.
- ^ а б Фарина, V .; Дж. Хим. Soc., 1991, 113, 9585-9595. (дои:10.1021 / ja00025a025 ).
- ^ а б http://hwpi.harvard.edu/files/myers/files/11-the_stille_reaction.pdf
- ^ Либескинд, Л.С .; Фенгл, Р.В. Дж. Орг. Хим., 1990, 55, 5359-5364. (дои:10.1021 / jo00306a012 ).
- ^ Фарина, V .; Кападия, С .; Бришнан, Б .; Ванг, С .; Либескинд, Л. Дж, Орг. Хим, 1994, 59, 5905-5911. (дои:10.1021 / jo00099a018 ).
- ^ а б Mee, S. P. H .; Ли, V .; Болдуин, Дж. Э. Angew. Хим. Int. Ред., 2004, 43, 1132-1136.
- ^ Либескинд, Л.С .; Пенья-Кабрера, Э. Органикалық синтез, Coll. Том. 10, с.9 (2004); Том. 77, с.135 (2000). (Мақала )
- ^ Скотт, В.Дж .; Стилл, Дж. К. Дж. Хим. Soc., 1986, 108, 3033-3040. (дои:10.1021 / ja00271a037 ).
- ^ Джонсон, К.Р .; Адамс, Дж. П .; Браун, М.П .; Сенанаяке, C. B. W. Тетраэдр Летт., 1992, 33, 919-922. (дои:10.1016 / S0040-4039 (00) 91576-4 )
- ^ Наир, V .; Тернер, Г.А .; Чемберлен, С. Дж. Хим. Soc., 1987, 109, 7223-7224. (дои:10.1021 / ja00257a071 ).
- ^ Джуссиум, Б .; Квон, В .; Верлхак, Дж.Б .; Денат, Ф .; Дубак, Дж. Синлетт, 1993, 117-118. (дои:10.1055 / с-1993-22368 )
- ^ Шефи, Ф. К .; Годшалькс, Дж. П .; Стилл, Дж. К. Дж. Хим. Soc., 1984, 106, 4833-4840. (дои:10.1021 / ja00329a032 )
- ^ http://www.sigmaaldrich.com/chemistry/chemistry-products.html?TablePage=16246425
- ^ Диетмар Сейферт (1959). «Di-n-бутилдивинилтин ». Org. Синт. 39: 10. дои:10.15227 / orgsyn.039.0010.
- ^ Қасқыр, С .; Леребурс, Р. Дж. Орг. Хим., 2003,68 7551-7554. (дои:10.1021 / jo0347056 ).
- ^ Қытырлақ, Г.Т .; Глинк, П. Т. Тетраэдр, 1994, 50, 2623. (дои:10.1016 / S0040-4020 (01) 86978-7 )
- ^ Бейли, Т.Р. Тетраэдр Летт., 1986, 27, 4407. (дои:10.1016 / S0040-4039 (00) 84964-3 ).
- ^ Бао, З .; Чан, В .; Ю, Л. Хим. Mater., 1993, 5, 2-3. (дои:10.1021 / cm00025a001 ).
- ^ Бао, З .; Чан, В.К .; Ю, Л. Дж. Хим. Soc., 1995, 117, 12426-12435. (дои:10.1021 / ja00155a007 ).
- ^ Sun, S. S .; Льюис, Дж. Э .; Чжан, Дж .; Цзян, Х .; Чжан, С .; Матос, Т .; Ли, Р .; Полим. Хим., 2010, 1, 663-669. (дои:10.1039 / B9PY00324J )
- ^ Лебсак, А. Д .; Сілтеме, Дж. Т .; Овермен, Л. Е .; Старнс, Б. Дж. Хим. Soc., 2002, 124, 9008-9009. (дои:10.1021 / ja0267425 )
- ^ Массе, C. Е .; Янг М .; Соломон Дж .; Панек, Дж. С. Дж. Хим. Soc., 1998, 120, 4123-4134. (дои:10.1021 / ja9743194 )
- ^ Мартин, Ф.; Хамфри, Дж. М .; Али, А .; Хиллиер, М. Дж. Хим. Soc., 1999, 121, 866-867. (дои:10.1021 / ja9829259 )
- ^ Кенде, А.С .; Кавамура, К .; Девита, Р. Дж. Дж. Хим. Soc., 1990, 112 4070-4072. (дои:10.1021 / ja00166a072 ).
- ^ Кенде, А.С., Кох, К .; Дори, Г .; Калдор, I .; Лю, К. Дж. Хим. Soc., 1993, 115, 9842-9843. (дои:10.1021 / ja00074a078 ).
- ^ Гонконг, C. Y, Киши, Y. Дж. Хим. Soc., 1991, 113, 9693-9694. (дои:10.1021 / ja00025a056 ).
- ^ Танимото, Н .; Герриц, С.В .; Савэбе, А .; Нода, Т .; Филла, С.А .; Масамуне, С. Angew. Хим. Int. Ред., 2003, 33, 673-675. (дои:10.1002 / ань.199406731 ).
- ^ Эванс, Д.А .; Қара, В. Дж. Хим. Soc., 1993, 115, 4497-4513. (дои:10.1021 / ja00064a011 ).
- ^ Тан, В .; Прусов, Е. В. Org. Летт., 2012, 14 4690-4693. (дои:10.1021 / ol302219x ).
- ^ Коулман, Р.С .; Вальчак, М .; Кэмпбелл, Э.Л. Дж. Хим. Soc., 2005, 127, 16036-16039. (дои:10.1021 / ja056217g ).
- ^ Рот, Г. П .; Фарина, V .; Либескинд, Л.С .; Пенья-Кабрера, Э. Тетраэдр Летт. 1995, 36, 2191.
- ^ Ренальдо, А.Ф .; Лабади, Дж. В .; Стилл, Дж. К. Органикалық синтез, Coll. Том. 8, б. 268 (1993); Том. 67, 86-бет (1989). (Мақала )
- ^ Төмен еритін қант-мочевина-тұз қоспаларында стетр реакциясы тетраалкилстаннандармен және фенилтриалкилстаннандармен.Джованни Императо, Рудольф Васольд, Бурхард Кёниг Жетілдірілген синтез және катализ 348 том, 15 басылым, 2243–47 беттер 2006 дои:10.1002 / adsc.2006
- ^ П. Эспинет, А.М. Эчаваррен (2004). «Стилл реакциясының механизмдері». Angewandte Chemie International Edition. 43 (36): 4704–4734. дои:10.1002 / anie.200300638. PMID 15366073.
- ^ Найт, С.Д .; Овермен, Л. Е .; Пайро, Г. Дж. Хим. Soc., 1993, 115, 9293-9294. (дои:10.1021 / ja00073a057 )
- ^ Монера, Э .; Ортар, Г. Биорг. Мед. Хим. Летт., 2000, 10, 1815-1818. (дои:10.1016 / S0960-894X (00) 00344-9 ).
- ^ Джоркос, А. С .; Стилл, Дж. К .; Хегедус, Л. Дж. Хим. Soc., 1990, 112, 8465-8472. (дои:10.1021 / ja00179a035 ).
- ^ Юэ, В.С .; Ли, Дж. Дж. Org. Летт., 2002, 4, 2201-2203. (дои:10.1021 / ol0260425 )
Сыртқы сілтемелер
- Стиль реакциясы туралы үлестірме Myers тобынан.
- Стилл реакциясы organic-chemistry.org сайтында
- Стиль реакциясы - синтетикалық хаттамалар organic-reaction.com сайтынан